Introduction
Caribou (
Rangifer tarandus) are distributed globally throughout Arctic, sub-Arctic, and boreal forest regions (
Banfield 1961). Predation, climate change, and habitat loss due to development and natural resource extraction have caused declines in population sizes across their range and continue to be major threats to their survival (
Festa-Bianchet et al. 2011). In Canada, several ecotypes of caribou have been identified based on morphology, ecology, and genetic differences (
COSEWIC 2002;
Klütsch et al. 2016) with further classification into designatable units (DUs) for conservation (
COSEWIC 2011). Within Canada, boreal caribou within central Ontario (also known as forest-dwelling woodland caribou) are listed as Threatened under the Federal
Species at Risk Act (
COSEWIC 2014) and under
Ontario’s Endangered Species Act, 2007.
Boreal woodland caribou typically select mature conifer stands where they can forage on ground lichen (
Cladina spp.) (
COSEWIC 2002;
McGreer et al. 2015;
McNeill et al. 2020). Lichen colonizes soil and land plants (
Favero-Longo and Piervittori 2010) and is formed through a symbiotic relationship between a fungus (mycobiont) and a population of algae (photobionts) (
Bates et al. 2011;
Banchi et al. 2018). Although traditionally thought to involve only a single fungal species, lichen is increasingly thought to include two or more species that make up the mycobiome of the lichen (
Spribille et al. 2016;
Banchi et al. 2018;
Tuovinen et al. 2019). Caribou are unusual in that they are one of only a few animals for which lichen typically makes up 46%–70% of their diet (
Thompson et al. 2015). Even in summer months when green plants are widely available, observational accounts suggest significant reliance of woodland caribou on lichen (
Thompson et al. 2015;
McNeill et al. 2020). However,
Denryter et al. (2017) suggested this reliance on lichen is an oversimplification and that studies of caribou diet are often limited by seasonal sampling and biases associated with traditional postingestion techniques such as analysis of rumen contents and faecal microhistology that can lead to poor taxonomic resolution. A combination of observational video, microhistological faecal preparations, and a DNA barcoding approach with cloning and Sanger sequencing suggests that DNA-based approaches provided substantially higher taxonomic resolution (94% identified to species) over video (42% identified to species) and histological approaches (<15% identified to species) (
Newmaster et al. 2013).
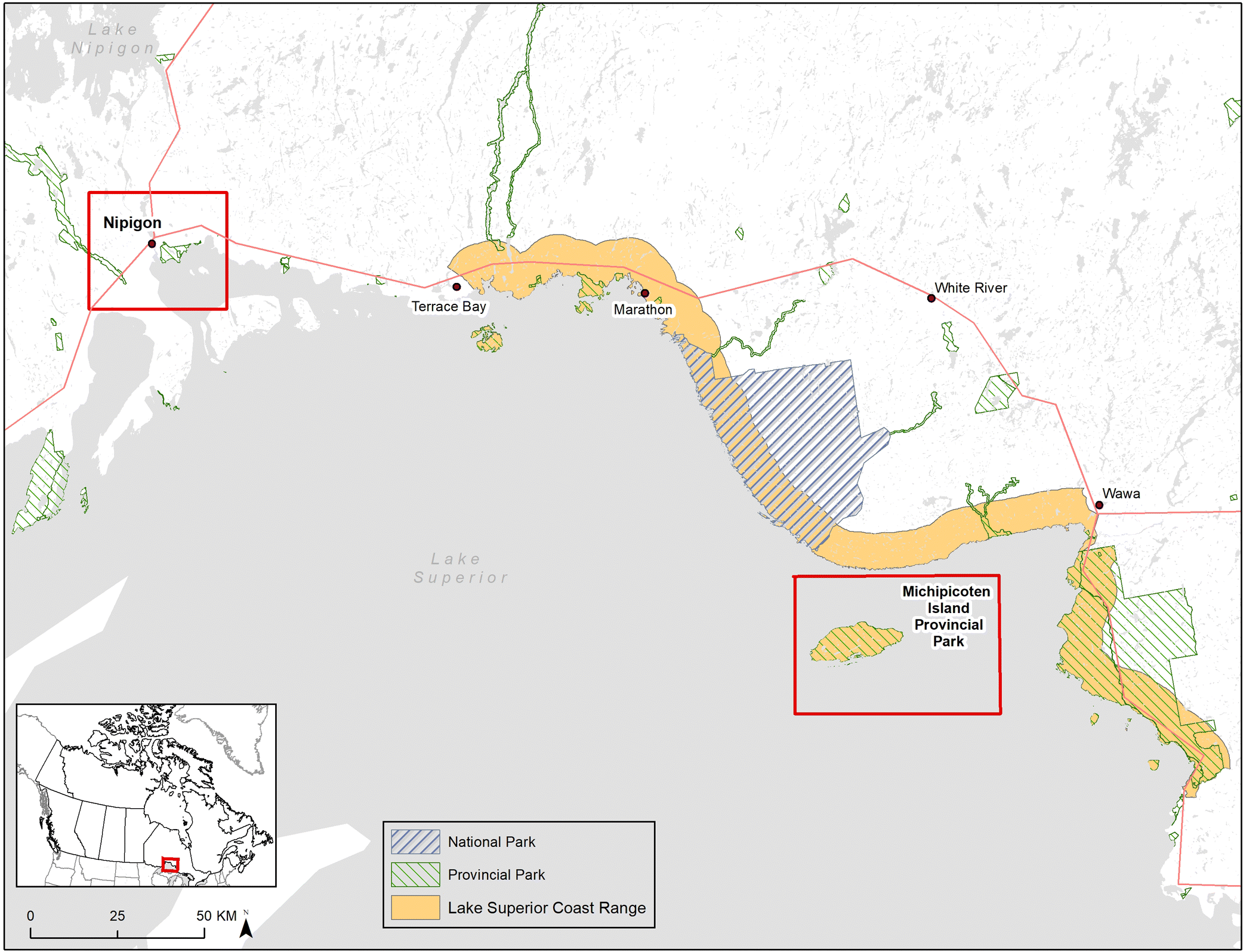
In Ontario, woodland caribou populations in the Lake Superior Coastal Range (
Fig. 1) were declining dramatically in the early 1980s, so in 1982 seven individuals (1 male, 3 adult females with 3 female calves) were translocated from the Slate Islands in northern Lake Superior to join a single adult male on Michipicoten Island (
Bergerud et al. 2007;
Drake et al. 2018), a large, predator-free island in Lake Superior (
Fig. 1). An additional male was added to the island in 1989 (
Bergerud 2007). The caribou population on Michipicoten Island likely numbered > 500 caribou by winter 2014 (
B.R. Patterson unpublished data). The vegetation on Michipicoten Island differs from that of the mainland in that lichen is very limited (
Bergerud et al. 2007). Observational accounts suggest that caribou on the island have a more deer-like diet that includes leafy material, twigs, and buds from deciduous trees and shrubs (B. R. Patterson, personal communication), but this has not been confirmed with any directed diet analysis. During 2014 and 2015, wolves crossed ice bridges that temporarily connected the island to the mainland and caribou became the primary food source for the colonizing wolves. As wolf numbers increased, caribou numbers decreased, and concerns were raised about the persistence of caribou on the island so some of the remaining caribou were translocated back to the Slate Islands and Caribou Island in the winter of 2018.
Caribou recovery strategies have mainly focused on predation and habitat loss (
Environment Canada 2012) with little research on diet diversity in environments with variable flora (but see also
McNeill et al. 2020 and references contained therein). Currently, the diet of caribou across its various ecotypes is not well understood (
Newmaster et al. 2013), but clarifying variable dietary habits is important for conservation and management policy to ensure responses such as translocation efforts are successful. This information will become increasingly important as lichen availability shifts in response to climate change and habitat alteration associated with development (
Allen et al. 2019).
The approach of metabarcoding environmental DNA (eDNA) followed by next-generation sequencing has recently become a favoured approach for species identification in environmental samples, because it provides a relatively fast and efficient way to obtain species-specific data by simultaneously sequencing DNA from complex mixtures. This method has been successful in a variety of environmental DNA studies, including diet analysis (
Berry et al. 2017), whereby DNA metabarcoding involves amplifying DNA from faeces with taxon-specific primers that target specific regions within the DNA (
Creer et al. 2016). With careful selection of molecular markers and access to sequence databases, forage species can be reliably identified in herbivores (
Kartzinel et al. 2015) and prey species can be revealed in the scat of carnivores (
Monterroso et al. 2019). Overall, this process allows for a relatively inexpensive approach to identify numerous target species within a single sample (
Fonseca 2018) and is quickly becoming the favoured approach to assess dietary habits of aquatic and terrestrial animals (
de Sousa et al. 2019).
To develop a better understanding of the potential for woodland caribou to adapt their diet to different environments, we used DNA metabarcoding and next-generation sequencing to analyze caribou scat samples collected in a lichen-rich environment on the mainland (Nipigon region) and in a lichen-poor environment on Michipicoten Island (
Fig. 1). We used two different markers, one to capture the diversity of green plants (
trnL region of the chloroplast genome) and another to identify lichen-associated fungal species (ITS2 region of the nuclear genome). Taxonomic identification of the lichen through amplification of the ITS2 region of the associated fungal species has been successful with previous Sanger sequencing methods (
Newmaster et al. 2013). Although caribou diet shifts seasonally (
Bergerud 1972;
Thompson et al. 2015;
Denryter et al. 2017), this research focuses on the winter diet due to the ease of scat collection in winter months and the availability of previously collected samples.
Materials and methods
Sampling, DNA extraction, and amplification
Archived caribou scat samples were used for this study. Samples were collected between February and March of 2007 from the Nipigon mainland region (
n = 10) and from Michipicoten Island on 27 February 2006 (
n = 10) (
Fig. 1) and stored at −20 °C. Although the Michipicoten samples were collected on a single day, they were collected by at least two different field biologists and each sample represents a different pellet cluster. Additional metadata (e.g., GPS coordinates) are unavailable for these archived samples. Additionally, scat samples from captive reindeer
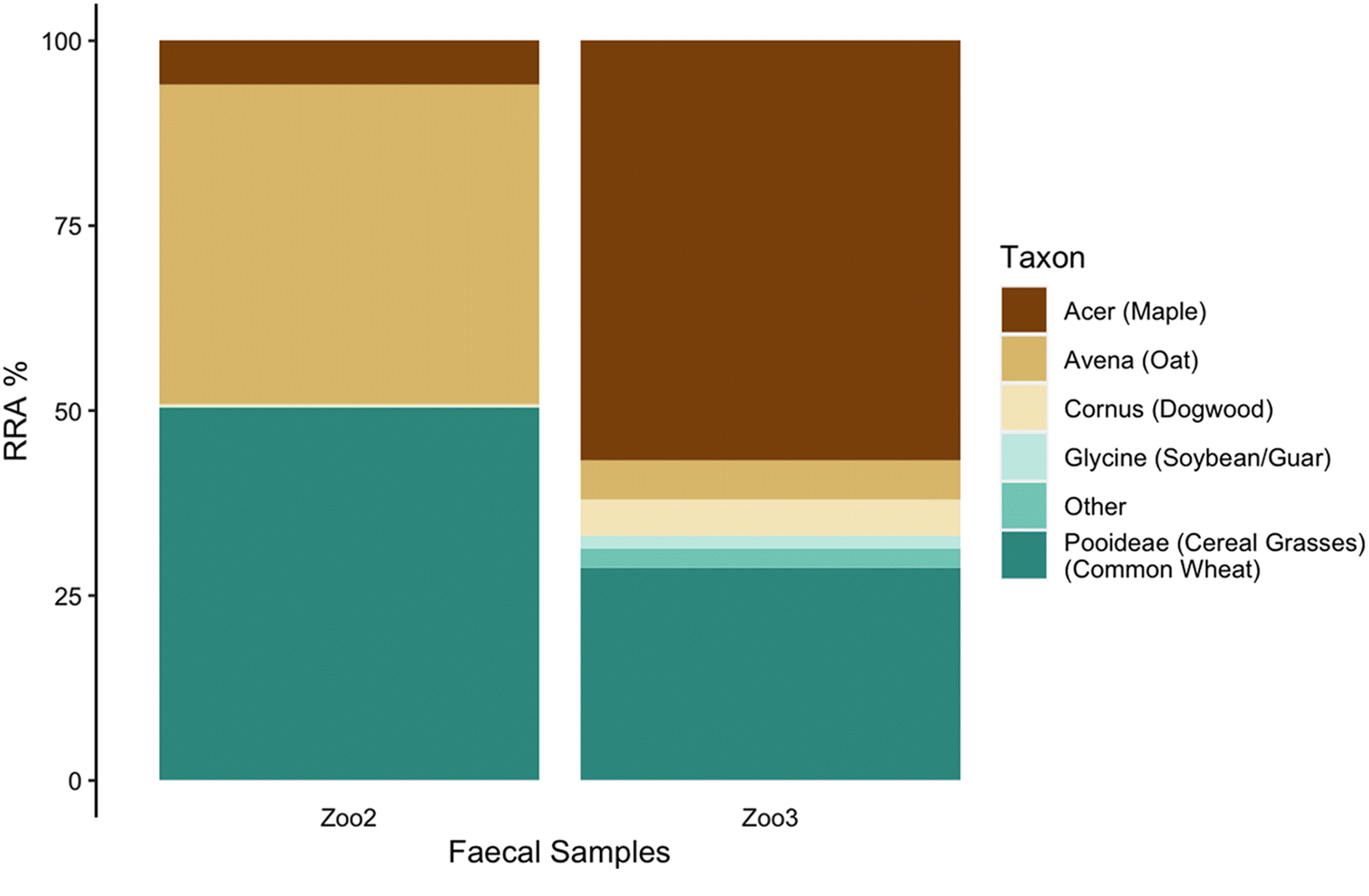
(Rangifer tarandus) were collected in September 2018 from the enclosure at the Peterborough Riverview Zoo as a positive control to validate our methods (
n = 3). Although visibly fresher samples were chosen from the reindeer enclosure, the actual age of each sample is unknown. Zoo reindeer were fed a diet of Zukudla Herbivore Pellets along with alfalfa, various browse (willow, dogwood, poplar, and maple) when available, and a small daily amount of apples and carrots (4 apples and 2 carrots) were made available. These animals are also free to eat available browse within the outdoor enclosure. The commercial pellets include (in order of abundance) wheat shorts, soybean meal, oat screenings, soy hulls, ground wheat, alfalfa, barley, beet pulp, ground flax, and wheat middlings. Although it is expected that the captive animals consumed primarily the pellets and woody browse, the specific daily relative consumption patterns of each item on any given day is not known.
For the samples collected from Nipigon and Michipicoten regions, two extraction replicates of 10 samples from each location were used with an additional two (trnL) or three (ITS2) positive control samples from the zoo. Faecal samples were prepared by thawing individual pellets, mashing within the collection bag, and then collecting approximately 50 mg per extraction into lysis buffer (Zymo Research Corporation, Irvine, CA). DNA was extracted with the Zymo Quick-DNA Faecal/Soil Microbe Kit (Zymo Research Corporation, Irvine, CA) according to manufacturer’s instructions. Negative controls were used throughout the extraction process to track any possible contamination. DNA from all samples was recovered by elution in a 100 μL volume of low TE (Tris-ethylenediaminetetraacetic acid (EDTA); 10 mM Tris pH 8, 0.1 mM EDTA).
Test amplification for both markers (
trnL and ITS2;
Table 1) was done on DNA from three zoo samples and three previously extracted water hyacinth (
Eichhornia crassipes) samples. Each polymerase chain reaction (PCR) used 1.0 mM PCR buffer (200 mM Tris-HCl, pH 8.4; 500 mM KCl), 1.5 mM MgCl
2, 0.2 mM dNTPs, 0.2 mM bovine serum albumin (BSA), 0.3 μM of each primer, 0.025 U of
Taq DNA polymerase, and 2 μL of DNA template in a final volume of 10 μL. Amplicons were visualized with gel electrophoresis on a 1.5% agarose gel containing Ethidium Bromide to ensure amplification of targeted size fragment.
Library preparation
To determine the optimal annealing temperatures, a temperature gradient PCR was conducted for both primer pairs using the following annealing temperatures; 63.8 °C, 59.1 °C, 55.7 °C, 63.8 °C and 51 °C. The 25 μL reaction consisted of 12.5 μL 2 × NEBNext Q5 Ultra II Q5 Master Mix (New England BioLabs, Whitby, ON, Cat No: M0543S), 0.3 μM of each primer (with Illumina overhang adaptor sequences (
Table 1)), 4 μL of DNA template and 7 μL molecular grade DNAase/RNAase free water. The amplification included four replicates of DNA from a zoo faecal sample (unquantified), and four replicates of DNA previously extracted from water hyacinth (standardized to 2 ng/μL) and a PCR negative. The reactions were carried out under the following conditions: initial denaturation at 98 °C for 30 seconds, denaturation at 98 °C for 10 seconds, annealing (63.8–51 °C) for 30 seconds and extension at 65 °C for 45 seconds, followed by 35 cycles and a final extension at 65 °C for 5 minutes. Based on these results, 62 °C was selected for future annealing temperature for the first stage PCR amplification in the library preparation protocol. All samples were independently amplified twice, and duplicate PCR products were pooled prior to indexing. Samples from both sampling locations and the zoo amplified at the ITS2 region, but samples from Nipigon did not amplify at the
trnL region and were therefore excluded from further analysis at this DNA marker.
Following the first-stage PCR, the pooled trnL amplicon PCR products were cleaned with 1.5X AMPure magnetic beads (Beckman Coulter, Mississauga) and the ITS2 amplicon PCR products were cleaned with 0.8X AMPure magnetic beads according to methods described in the 16S Illumina Metagenomic Sequencing Library Preparation protocol. The cleaned amplicon PCR product was visualized with an Agilent Tape Station 4200 (Agilent Technologies, Mississauga) with the D1000 reagent kit according to manufacturer’s directions to ensure amplification of target fragments. A second PCR was used to attach unique Nextera XT Dual Indices to our amplicon product (Nextera XT 24 Index kit; Illumina, Cat No. FC-131-1001) according to manufacturer’s instructions. Briefly, we used 25 μL of the 2x NEBNext Q5 Ultra II Q5 Master Mix, 5 μL of each Nextera XT index primers, 10 μL of DNase/RNase free molecular grade water and 5 μL of the amplicon DNA template. The index PCR included 10 cycles with the following conditions: initial denaturation at 98 °C for 30 seconds, denaturation at 98 °C for 10 seconds, annealing/extension at 65 °C for 75 seconds followed by final extension at 65 °C for 5 minutes. A second bead clean-up was done on the indexed PCR product with the same reagents and protocol as the first PCR bead clean-up with the exception that we used 1.12X (56 μL) of AMPure magnetic beads to capture our target fragment. Cleaned product was quantified with the High Sensitivity assay on a Qubit Fluorometer (Thermo-Fisher Scientific, Markham). Sample concentration was converted to nM and samples were standardized to 4 nM with elution buffer (EB; 10 mM Tris-Cl, pH 8.5) (Qiagen, Toronto). Samples at 4 nM were pooled (5 μL each) and the final library was visualized on the Agilent 4200 Tape Station and quantified with Qubit Fluorometer. In summary, 5 μL of all the Michipicoten (n = 10), Nipigon (n = 10), and zoo (n = 3) ITS2 samples were combined to form a single library (n = 23) and 5 uL of the Michipicoten (n = 10) and zoo (n = 2) trnL samples were combined into a second library (n = 12) for independent sequencing.
Sequencing, classification, and analysis
Pooled libraries were diluted to 6 pM and denatured with sodium hydroxide (NaOH) diluted with hybridizing buffer followed by heat denaturation according to Illumina sequencing instructions. Sequencing of the ITS2 library was completed with the MiSeq Reagent Nano Kit v2 (500 cycles; MS-103-1003), and for the trnL library we used the MiSeq Reagent Nano Kit v2 (300 cycles; MS-103-1002) with 2 million paired-end reads expected for each library. We included 30% PhiX (PhiX control kit v3, FC-110-3001) in each sequencing run to serve as an internal control and to ensure sufficient clustering on the flow cell when low diversity libraries are sequenced.
Sequences were automatically de-multiplexed on the MiSeq BaseSpace platform, and sequences were identified to species or genus following an adapted Metagenomics protocol in Geneious v 11.1.4 (Biomatters Ltd.). Briefly, reads were paired and trimmed to remove remaining Illumina adaptors, bases with a quality score below 30 and reads less than 100 bp (ITS) or 80 bp (trnL) were removed. Paired reads were merged to create a single consensus sequence. We then used the de novo assembly to cluster similar reads into operational taxonomic units (OTUs). We used a maximum mismatch per read of 2%, maximum per read gap of 1%, and minimum overlap identify of 98%. To create an ITS2 fungal taxonomy database locally within Geneious we: (i) downloaded the NCBI ITS project database (Fungi RefSeq ITS), (ii) uploaded it into Geneious, (iii) batch blasted the OTU consensus sequences against the Fungi RefSeq ITS database within Geneious with a maximum hit of 1 and scoring mismatch of 1–2, (iv) removed duplicates, (v) downloaded the hits list, and (vi) extracted the BLAST hit regions to create a database for sequence classification. We then classified the ITS2 amplicons with Geneious Sequence Classifier plug-in with high sensitivity and minimum overlap of 95%. We summarized the hits based on sequences that could be classified to the genus level, and then combined hits for each of Michipicoten, Nipigon, and zoo samples. Taxa that had fewer than 20 total reads were excluded. The process for classifying the trnL sequences was similar, except that initial BLAST of OTU consensus sequences was done within the online NCBI nucleotide database, and samples only include Michipicoten and two zoo samples because amplification at this marker was unsuccessful on the Nipigon samples. The third zoo sample (Sample Zoo1) was not included in the trnL library due to limited availability of DNA for the that sample. Only sequences that were at least 80 bp in length and had 100% identity based on BLAST results were included in the trnL summaries.
Relative read abundance was calculated for each detected item (RRA
i) based on sequence counts for both markers according to the following formula (
Deagle et al. 2019):
where
S is the number of samples,
T is the number of taxa, and
ni,k is the number of sequences for item
i in sample
k. To explore consistency of plant taxa detected across samples, we used
ggplot2 (
Wickham 2016) within the R (
R Core Team 2020) to create stacked bar charts of RRA for individual samples from Michipicoten and the zoo samples.
Discussion
Here, we demonstrate an optimized eDNA metabarcoding approach for taxonomic classification of plants and lichen in >10-year-old caribou faecal pellets. We found that metabarcoding and sequencing amplicons at the ITS2 region was useful for identifying lichen-associated fungi, although the methodology is limited by the reference samples in the fungal NCBI database. Similarly, DNA metabarcoding and sequencing of amplicons from the trnL chloroplast region provided reliable identification of plants at the genus (and sometimes family) level.
Lichen-associated fungus (
Lichenoconium aeruginosum) was evident in both wild populations, but the majority of fungal species identified are not known to be associated with lichen. These are presumably from alternate environmental sources. The identification of lichen-associated fungi in the Michipicoten samples suggests that caribou do have access to at least some terrestrial lichen on the island. Fungal species within the
Lichenoconium genus, including
L. aeruginosum, are known to associate with
Cladonia spp. of lichen (
Jando et al. 2000;
Zhurbenko and Pino-Bodas 2017), the predominant terrestrial lichen available to woodland caribou in Ontario (
Newmaster et al. 2013). Whether or not caribou will access lichen sources in their most recent translocation environment back on the Slate Islands, where arboreal lichen sources are more readily available than terrestrial sources (
Bergerud et al. 2007), will require sampling and analysis of caribou faecal pellets from that location. The high percentage of unclassified reads suggests that identification may be limited by the NCBI ITS2 fungal reference database and so a targeted effort to sequence the ITS2 region of various arboreal and terrestrial lichen could improve the database and potentially increase species assignments.
Although the trnL region only amplified in zoo and Michipicoten samples, the results provide support for our methodology and insight into the reliance of island caribou on alternative plant sources. In the zoo samples, the predominance of sequence reads from the three main ingredients in the commercial pellets (wheat, oats, soybean) and the supplemental woody browse (maple and dogwood) provide support that sequence reads reflect relative abundance, at least in broad terms. Although not every item listed in the commercial pellets nor every supplemental item made available was detected in the zoo samples, it is possible that items not detected were in small quantities in the pellets and (or) that high consumption of woody browse (e.g., maple) on any given day may have impacted the relative abundance in the faecal pellets collected. Alternatively, the parameterization and thresholds set for analysis may also have contributed to lack of detection of food items consumed in small quantities.
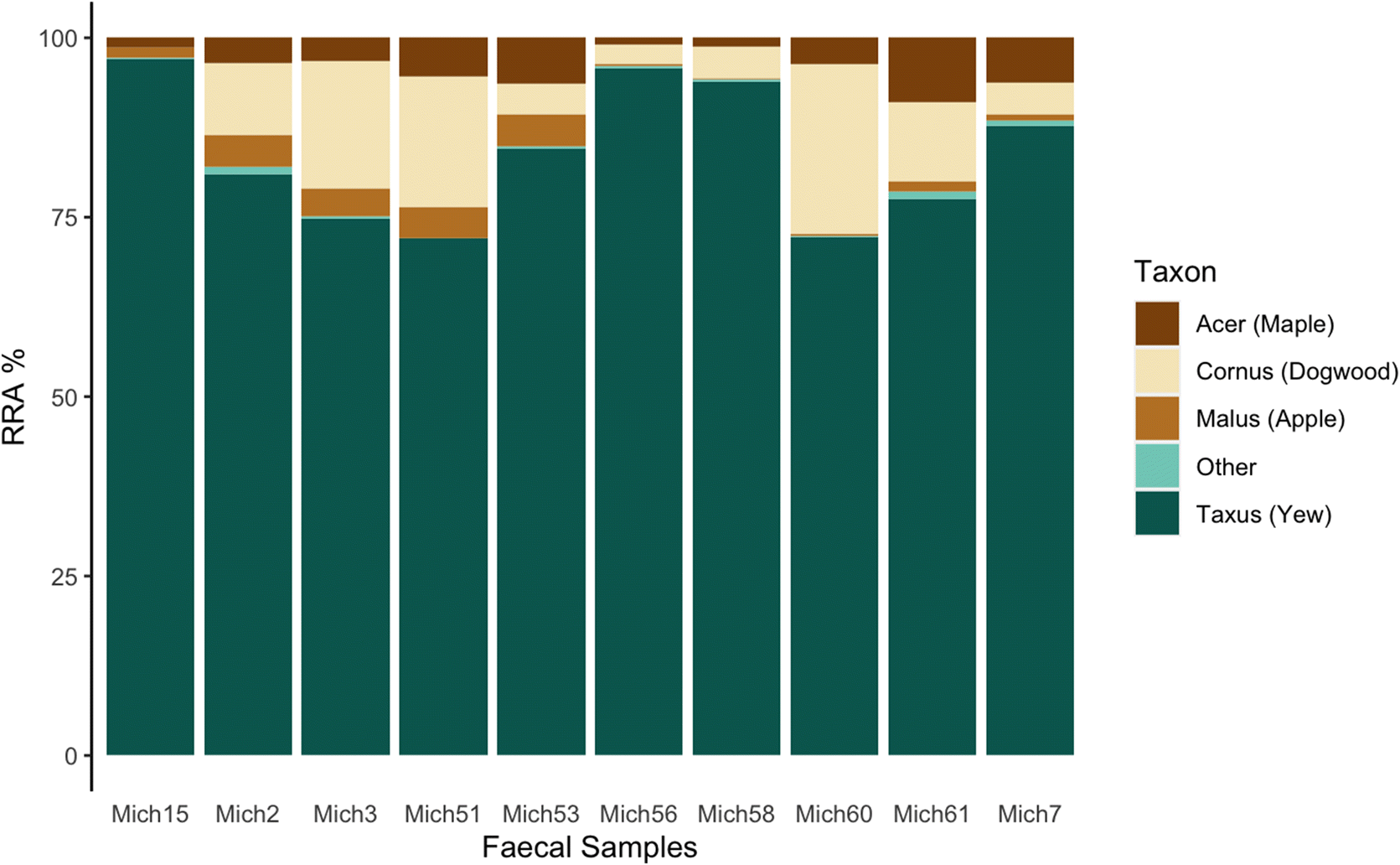
Woodland caribou are often considered to be obligate lichen foragers. It may seem surprising then that all individual Michipicoten samples had yew as the predominant plant consumed, and that on average yew accounted for >80% of the sequences, since
Taxus spp. contain relatively high concentrations of taxine alkaloids and are generally toxic to domestic grazers (reviewed by
Wilson et al. 2001 and
Windels and Flaspohler 2011). Wild cervids, however, readily browse on yew, and toxic effects have rarely been documented (e.g.,
Handeland 2008). Also, a decline in yew is often associated with overgrazing by ungulates. For example, the disappearance of Canada yew from Isle Royale within 2 decades of the arrival of moose in 1909 is well documented (
Murie 1934;
Snyder and Janke 1976). Similarly, Canada yew was considered functionally extirpated from the Slate Islands by 1985 (
Bergerud et al. 2007), putatively due to overbrowsing by caribou. In contrast, the high consumption of yew found in our study is consistent with a forage survey conducted on Michipicoten in 2011 that determined yew to be the dominant available forage for caribou despite almost three decades of increasing caribou abundance on the island (
Kuchta 2012).
The heavy consumption of yew by Michipicoten Island caribou has been noted in historical observational accounts (Eason pers. Comm. Cited in
Cumming 1992), but unconfirmed until now. Although yew is abundant on Michipicoten, it is rare in more northern regions of Ontario (
Cumming 1992), making it an unlikely food source in more northern landscapes. The near-extirpation of yew from the Slate Islands, and the fact that caribou were known to be threatened by food limitations there in the past (
Bergerud et al. 2007), may impact the fitness of caribou that have been recently translocated back there. Green forage plants provide higher protein (>30%) compared to terrestrial lichens (<4%) and should be preferred when available, but lichens are known to be important year-round even when alternatives are available (
Thompson et al. 2015). Continued monitoring of the diet and fitness of caribou in different environments with variable browse will provide important information on dietary shifts of caribou in response to environmental changes and food accessibility.
The ability of DNA metabarcoding to correlate sequence reads with proportional consumption has been controversial. Issues surrounding variable DNA copies across tissue sources, DNA degradation, and potential preferential binding of primers to specific species template have been presented as challenges to proportional consumption based on sequence read frequency (
Pompanon et al. 2012). However,
Kartzinel et al. (2015) found that mean sequence RRA of
trnL sequences was a reliable predictor of proportional consumption based on correlation with stable isotope analysis, although they focus on coarse taxonomic differentiation of plant families. The main food items identified in our
trnL analysis were yew (Taxacea:
Taxus), dogwood (Sapindaceae:
Cornus), maple (Cornales:
Acer), and apple (Rosaceae:
Malus), which all belong to different plant families, and therefore most likely reflect some relative level of biomass. Furthermore,
Deagle et al. (2019) reviewed the topic of interpreting sequence counts and conducted simulations to conclude that, in general, relative read abundance provides a more accurate view of diet analysis than does frequency of occurrence, particularly when the same food taxa occur across many samples. They also noted that sequence read counts are most reliable for population-level diet analysis where common items are represented by the highest number of reads. Our
trnL data reveal yew as the highest RRA in every sample, suggesting this species made up the bulk of caribou plant diet at that particular time. Furthermore, the results from the zoo samples, while imperfect and not specifically designed as a controlled feeding trial, do reflect the major food items made available. Therefore, it seems a reasonable conclusion that the high proportion of yew sequences found in our samples is representative of relative population-level consumption during winter.
We faced two main challenges with this work. First, the ITS2 analysis was limited by the reference sequences of the fungal NCBI database. Database coverage is often a limiting factor in DNA metabarcoding studies (
Pompanon et al. 2012), especially for uncommon sequencing regions like ITS2 and
trnL. The most common databases utilize mitochondrial DNA markers, especially cytochrome oxidase I (e.g., Barcode of Life Data (BOLD; boldsystems.org) developed at the Centre for Biodiversity Genomics in Canada, and the International Barcode of Life (iBOL; ibol.org), which are often not useful for dietary analysis where alternate markers are often used. In these cases, making a customized database may be required to fully disentangle dietary niches (
Deagle et al. 2009;
Rayé et al. 2011). To more thoroughly assess lichen diversity and alternative plant sources in caribou diet, we recommend collection of lichen and flora samples from faecal collection sites to create independent and comprehensive databases for regional lichen (ITS2) and plant (
trnL) sequences. In doing so, future work will be able to more accurately assess dietary needs of caribou by identifying lichen-associated fungi and plant sequences more reliably to the species level.
Second, amplification at the
trnL region failed in the mainland Nipigon samples, even though the samples were of similar age to the Michipicoten samples where amplification was successful. Degradation of DNA can be problematic in diet analysis studies and is often the cause of failed amplification (
Deagle et al. 2006). However, the
trnL primers amplify a chloroplast fragment that is about half the size of the nuclear ITS2 fragment we successfully amplified in all samples, which suggests DNA was not degraded in the Nipigon samples. Also, the
trnL primers amplify a wide variety of plant species (
Taberlet et al. 2007), so it is unlikely that mismatched primer binding sites were the reason for failed amplification. Therefore, the failed amplification may truly be reflective of the late winter diet of mainland caribou in which they rely heavily, and possibly exclusively, on lichen in regions where it is abundant and accessible (
Thompson et al. 2015). Further exploration with larger sample sizes and broader temporal sampling would help corroborate our results.
Maintaining and stabilizing woodland caribou populations is a key objective for caribou conservation and recovery in Canada (
Environment Canada 2012). Predation, industrial development, and climate change have been identified as the major threats to caribou persistence (
Festa-Bianchet et al. 2011), but the variability in food resources and bottom-up regulation has received limited recent attention for woodland caribou populations (but see
Bergerud 1972,
1996;
Newmaster et al. 2013). Although lichen is recognized as a critical winter food source for caribou, the dependence on alternative dietary sources for woodland caribou in lichen-poor environments has not been fully recognized. Our results suggest that yew (
Taxus spp.) provides a substantial proportion of dietary needs to caribou on Michipicoten Island and may be the preferred browse when lichen is less available. We recognize, however, that interpretation is limited by small sample sizes and that our results do not exclude the possibility that other species (e.g., dogwood, maple) could be heavily targeted in the absence of yew (
Bergerud 1996). We also recognize that it is currently unknown if yew contributes significantly to the overall caloric and nutritional requirements of caribou or if it serves primarily the needs of the gut microbiome. Regardless, our results are important for conservation efforts that include translocation of animals to environments with variable flora, or where yew or other woody browse is inaccessible or overgrazed, such as the Slate Islands (
Bergerud 1996). Although the current lack of wolves on the Slate Islands will limit top-down regulation, the lack of yew and maple as an alternative food source could impact caribou viability through bottom-up limitations (
Bergerud et al. 2007;
Thompson et al. 2015). Although caribou diet may be region specific, with different food resources supporting different caribou populations, the nutrient availability in the variable forage will also need to be considered.
Overall, our results provide preliminary eDNA data that corroborates historical accounts of the diet versatility of caribou (
Bergerud 1972) and flags the importance of variable food resources to caribou persistence. Furthermore, we document a reliable methodology that can be applied to historical/archived samples to support caribou conservation on the Slate Islands, but more broadly to ungulate species across more extensive spatial and temporal scales.