An examination of the Iridovirus core genes for reconstructing Ranavirus phylogenies
Abstract
Ranaviruses are globally emerging infections of poikilothermic vertebrates and belong to the viral family Iridoviridae. The six species of ranaviruses are responsible for unknown numbers of infections and disease and mortality events around the world in amphibians, fish, and reptiles. Genomic investigations have shown that there are 24 core genes shared by all iridoviruses. In this study, we examine the utility of each of these genes in reconstructing phylogenetic relationships across six species of Ranavirus. We also performed dot-plot analysis for the 17 isolates in the study. For large-scale differentiation, using the major capsid protein gene creates a tree similar to the whole genome tree. Other comparable genes include open reading frame (ORF) 19R (a serine–theonine protein kinase) and ORF 88R (Erv I/Alr Family protein). The poorest candidate for phylogenetic reconstruction, due to high homology, was ORF 1R (a putative replication factor and (or) DNA binding-packing protein). There are a plethora of genes that may be useful to examine phylogenies at smaller scales (e.g., to examine local adaptation); however, they do not necessarily belong to the set of highly conserved core genes.
Introduction
Ranaviruses from the viral family Iridoviridae are large double-stranded DNA viruses with a large vertebrate host range (Duffus et al. 2015; Chinchar et al. 2017). They were first described in North American northern leopard frogs (Rana pipiens now Lithobates pipiens) in the mid-1960s (Granoff et al. 1965). Since then, they have been described in over a hundred other species of amphibians, reptiles, and fish from all around the globe (Duffus et al. 2015). Ranaviruses are responsible for countless morbidity and mortality events in affected species and have even caused population declines (e.g., common frogs (Rana temporaria) in the UK; Teacher et al. 2010) and local extirpations (e.g., multiple species in the Spanish Pyrenees; Price et al. 2014). They are also known to affect threatened and endangered species such as the Chinese giant salamander (Andrias davidanus, Geng et al. 2011) and pallid sturgeon (Scaphirhynchus albus, Waltzek et al. 2014), making them a cause for conservation concern.
Currently, there are six species of ranavirus recognized by the International Committee on Taxonomy of Viruses (ICTV; Chinchar et al. 2017); this is as a result of a deeper understanding of Ranavirus genomics. Initially, it was thought that the major capsid protein (MCP) was adequate for the study of viral evolution of iridoviruses, as it has both highly conserved, but also some variable domains (Tidona et al. 1998). However, some studies have used only portions of the MCP and have had trouble parsing possible local adaptations (e.g., Duffus and Andrews 2013). Other studies have used multiple genes, including the MCP, and have been able to detect genetic variation in large-scale data sets (e.g., Stöhr et al. 2015) and in genetic information from geographically isolated data sets (Ridenhour and Storfer 2008). While the MCP gene may have enough variation to place ranaviruses into their different species, a study that uses only the MCP may miss local variation or adaptation. For example, Ridenhour and Storfer (2008) only found evidence of local adaptation of Ambystoma tigrinum virus strains in the southeastern United States when they examined >5% of the genome and included genes such as the eIF-2α homologue, which is not present in all iridoviruses.
Eaton et al. (2007) reexamined the genomes of 12 different iridoviruses. They found that there were 26 core genes across all iridoviruses. These core genes are typically conserved, with functions that are generally associated with virulence, replication, and gene expression (Eaton et al. 2007; Jancovich et al. 2015). However, further molecular studies of newly discovered and sequenced iridovirids bring this total of shared genes down to 24 (e.g., Shrimp hemocyte iridescent virus lacks the small subunit of ribonucleotide reductase (Qiu et al. 2018) and European Chub Iridovirus lacks a deoxinucleotide reductase (GenBank accession number MK6376310)). Here we examine the utility of 24 core iridovirus genes across six species of ranavirus and perform a large scale dot-plot analysis of 17 different isolates. However, it is important to note that all ranaviruses contain the original 26 core genes and a number of other genes that are found in all ranaviruses. We hope that the results from this study will be useful for those who study ranavirus phylogenomics, genomics, and phylogenetics.
Materials and methods
Sequence data and initial analysis
Sequence data from 24 iridovirus core genes were obtained from GeneBank. The accession numbers of all isolates used, their abbreviations used for this study, and the isolate name as it appears in GenBank can be found in Table 1. Also see Table 1 for strains that are representative of each species of ranavirus. Sequence data for each gene was aligned using the default settings of the MAFFT server (Katoh et al. 2019; mafft.cbrc.jp/alignment/server/). FASTA formatted text files from the MAFFT alignment were then converted to MEGA format and were then analyzed to find the best fit nucleotide substitution model (Table 2).
Table 1.
| Species designation | Name | Abbreviation | GenBank accession number |
|---|---|---|---|
| Ambystoma tigrinum virus (ATV) | Ambystoma tigrinum virus—RRV | ATV-3 | KR075879.1 |
| Ambystoma tigrinum virus—UTAH | ATV-2 | KR075877.1 | |
| Ambystoma tigrinum virus | ATV-1 | NC_005832.1 | |
Chinese Giant Salamander Virus (CGSV) | Andrias davidianus ranavirus—2010SX | CGSV-1 | KF033124.1 |
| Andrias davidianus ranavirus—1201 | CGSV-2 | KC865735.1 | |
Chinese giant salamander iridovirus isolate CGSIV-HN1104 | CGSV-3 | KF512820.1 | |
| Common midwife toad virus (CMTV) | Common midwife toad ranavirus—Mesotriton alpestris/2008/E | CMTV-1 | JQ231222.1 |
Common midwife toad ranavirus isolate Pelophylax kl. esculentus/2013/NL | CMTV-2 | KP056312.1 | |
Common midwife toad ranavirus isolate Pe/2016/Netherlands/UU3160714042 | CMTV-3 | MF125270.1 | |
Epizootic haematopoietic necrosis virus (EHNV) | Epizootic haematopoietic necrosis virus | EHNV-1 | FJ433873.1 |
| European catfish virus (ECV) | European catfish virus | ECV-1 | KT989884.1 |
| European catfish virus | ECV-2 | KT989885.1 | |
| European sheatfish virus (ESV) | European sheatfish virus | ESV-1 | JQ724856.1 |
| Frog virus 3 (FV3) | Frog virus 3 | FV3-1 | NC_005946.1 |
| Frog virus 3 isolate SSME | FV3-2 | KJ175144.1 | |
| Bohle iridovirus | BIV-1 | KX185156.1 | |
| Singapore grouper iridovirus (SGIV) | Singapore grouper iridovirus | SGIV-1 | NC_006549.1 |
Table 2.
| FV3 ORF | Gene namea | MEGA 6 best fit model | Distance between MCP treeb | Distance between full genome treec |
|---|---|---|---|---|
| ORF 1R | Putative replication factor and (or) DNA binding or packing protein | Kimura Two Parameter and Invariability | 12 | 14 |
| ORF 2L | Myristilated membrane protein | Kimura Two Parameter and Invariability | 16 | 14 |
| ORF 9L | Putative NTPase I | Kimura Two Parameter and Gamma Distributed | 14 | 16 |
| ORF 12L | Unknown | Kimura Two Parameter and Invariability | 10 | 14 |
| ORF 15R | ATPase-like Protein | Kimura Two Parameter | 14 | 18 |
| ORF 19R | Serine–threonine protein kinase | Kimura Two Parameter | 10 | 12 |
| ORF 21L | Helicase family | Kimura Two Parameter | 16 | 16 |
| ORF 22R | D5 family NTPase involved in DNA replication | Kimura Two Parameter | 14 | 18 |
| ORF 37R | NIF–NLI interacting factor | Kimura Two Parameter | 12 | 16 |
| ORF 41R | Unknown | Kimura Two Parameter | 22 | 22 |
| ORF 53R | Myristilated membrane protein | Kimura Two Parameter | 22 | 22 |
| ORF 57R | Serine–threonine protein kinase | Kimura Two Parameter | 14 | 18 |
| ORF 60R | DNA polymerase family B exonucleases | Kimura Two Parameter | 10 | 12 |
| ORF 62L | DNA-dependent RNA polymerase II second largest subunit | Kimura Two Parameter | 10 | 14 |
| ORF 67L | Ribonucleotide reductase small subunit | Kimura Two Parameter | 6 | 14 |
| ORF 80L | Ribonuclease III | Kimura Two Parameter | 4 | 14 |
| ORF 81R | Transcription elongation factor TFIIS | Kimura Two Parameter | 8 | 20 |
| ORF 84R | Proliferating cell nuclear antigen | Kimura Two Parameter | 14 | 14 |
| ORF 85R | Deoxynucleoside kinase | Kimura Two Parameter | 10 | 14 |
| ORF 88R | Ervl/Alr family | Kimura Two Parameter | 12 | 14 |
| ORF 90R | Major capsid protein | Kimura Two Parameter | 0 | 12 |
| ORF 91R | Immediate early protein infected-cell protein-46 | Kimura Two Parameter | 10 | 10 |
| ORF 94L | Hypothetical protein—Clostridium tetani | Kimura Two Parameter | 6 | 10 |
| ORF 95R | Putative XPPG-RAD2-type nuclease | Kimura Two Parameter | 14 | 8 |
aGene name as defined by Eaton et al. (2007).
bDistance between the major capsid protein (MCP) tree and the stated tree in CompPhy (Fiorini et al. 2014) as calculated by Treedist based on the symmetric distance (Felsenstein 2008).
cDistance between the full genome sequence tree and the stated tree in CompPhy (Fiorini et al. 2014) as calculated by Treedist based on the symmetric distance (Felsenstein 2008).
Phylogenetic analysis
Phylogenetic analysis was done in MEGA 6 (Tamura et al. 2013). Maximum likelihood trees were built using the best fit nucleotide substitution model (Table 2; Figs. S1–S22). When the full genome trees were produced, noncollinear segments were left as such and no attempts at genome reorganization were undertaken. (This does not appear to have affected our results, because the trees share similar topologies with those created with the previously described 26 iridovirid core genes by Eaton et al. (2007) (e.g., Eaton et al. 2010; Jancovich et al. 2015)). With the increasing access to full genome sequencing, the use of the concatenated core genes will become less common and using full genomes will become the norm, and adjustments for noncolinear segments of the genome will less often be corrected. Open reading frame (ORF) 8R (partially duplicated gene in Singapore grouper iridovirus (SGIV)) or ORF 27R (truncated gene in SGIV) in our analyses of ranaviruses were not included in our analysis (data not shown). This is the reason why there are 24 genes in the analysis. The trees that we created are also unrooted.
Trees were compared visually using bootstrap values and distances (provided by the scale bars). To quantify the differences between the full genome tree, the MCP tree and the trees made from each ORF, CompPhy (Fiorini et al. 2014) was used, and Treedist was then used to determine the symmetrical distance between each ORF tree and the full genome or MCP (Felsenstein 2008) (Table 2). Trees that had a symmetrical distance of 10 and under were considered to be adequate for phylogenetic reconstruction.
Dot-plot analysis was performed in R (R Core Team 2019) in the package DICIPHER v 2.6 (Wright 2016).
Results
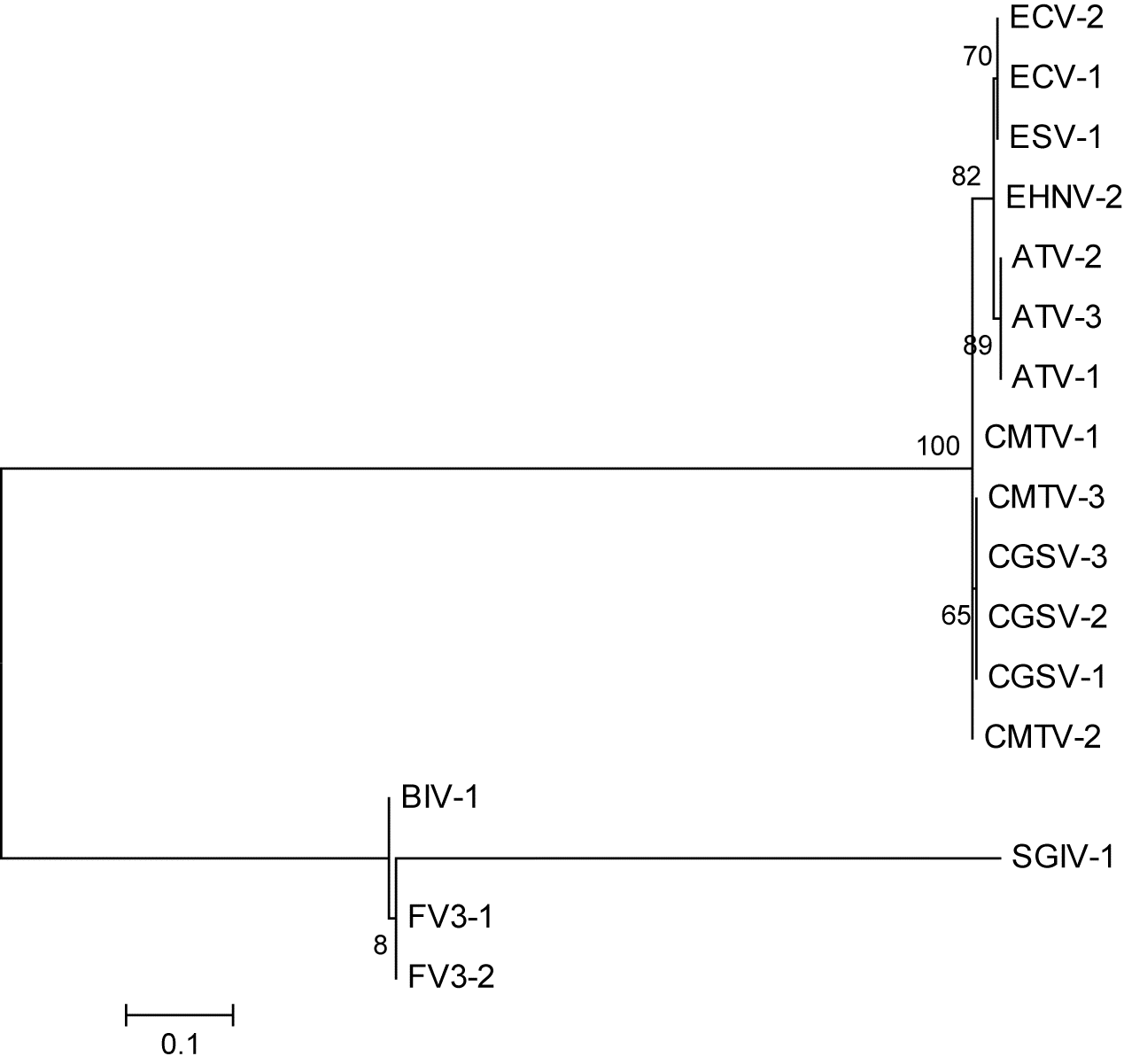
Most of the phylogenetic trees for the 24 core genes of the 17 isolates of ranavirus that were examined were able to group at the species level (six species). SGIV, however, is problematic as it appears as an individual group. It is important to note that at the time of this analysis only one sequence of SGIV was available; however, Grouper Iridovirus (not included in this analysis) may be a strain of SGIV. The tree constructed with full genome sequences (Fig. 1) shows the usual groupings of the six ranavirus species used in this study. Visually, the best trees, besides those made by the major capsid protein (ORF 90R in Frog virus 3 (FV3); Fig. 2), are ORF 19R in FV3 (Fig. 3), and ORF 88R in FV3 (Fig. 4). One of the worst ORFs for reconstruction was ORF 1R in FV3 (Fig. S1) because the branch lengths were extremely short between the different species with low bootstrap support. (All other trees can be found in the Figs. S2–S22.)
Fig. 1.
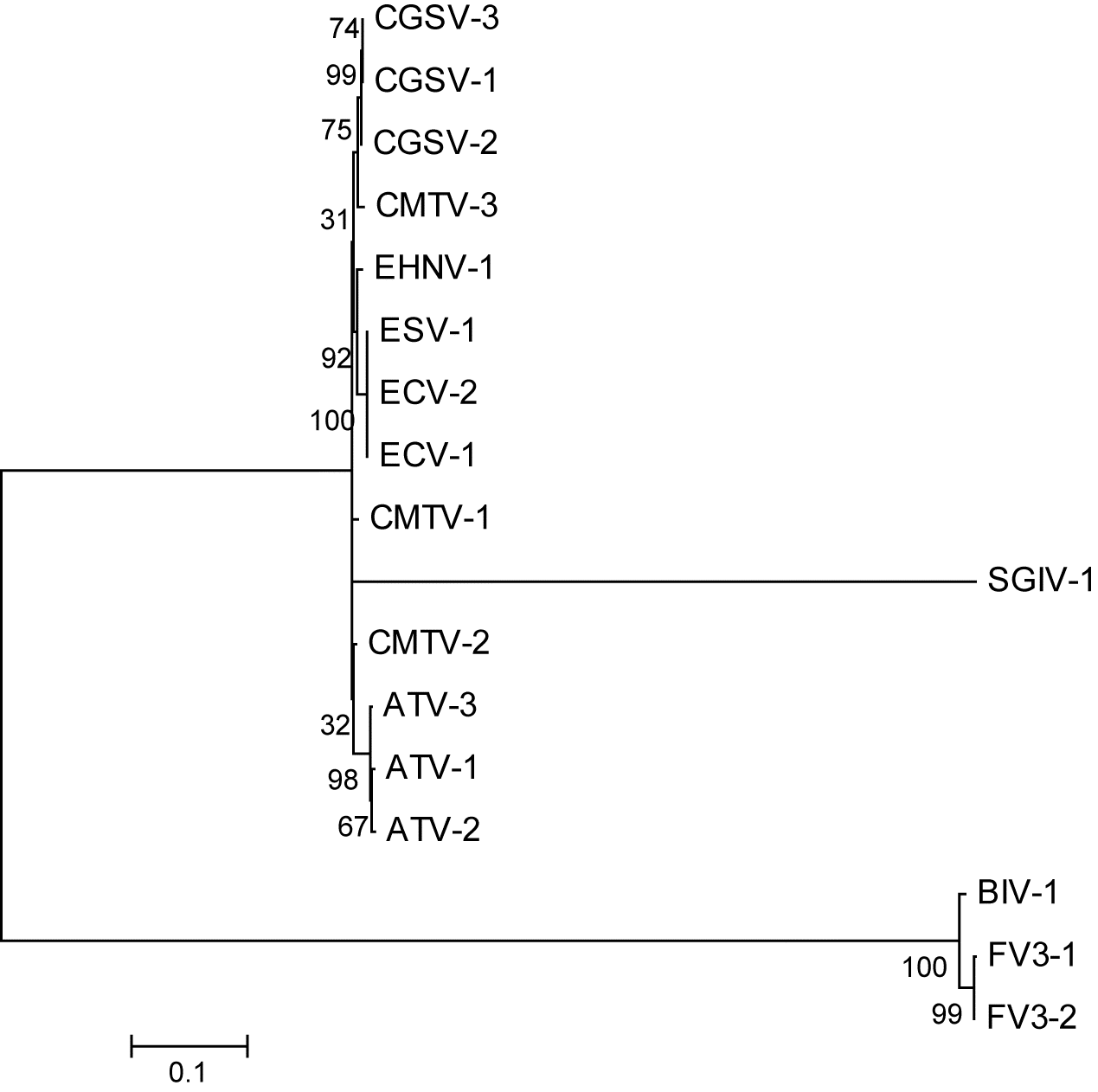
Fig. 2.
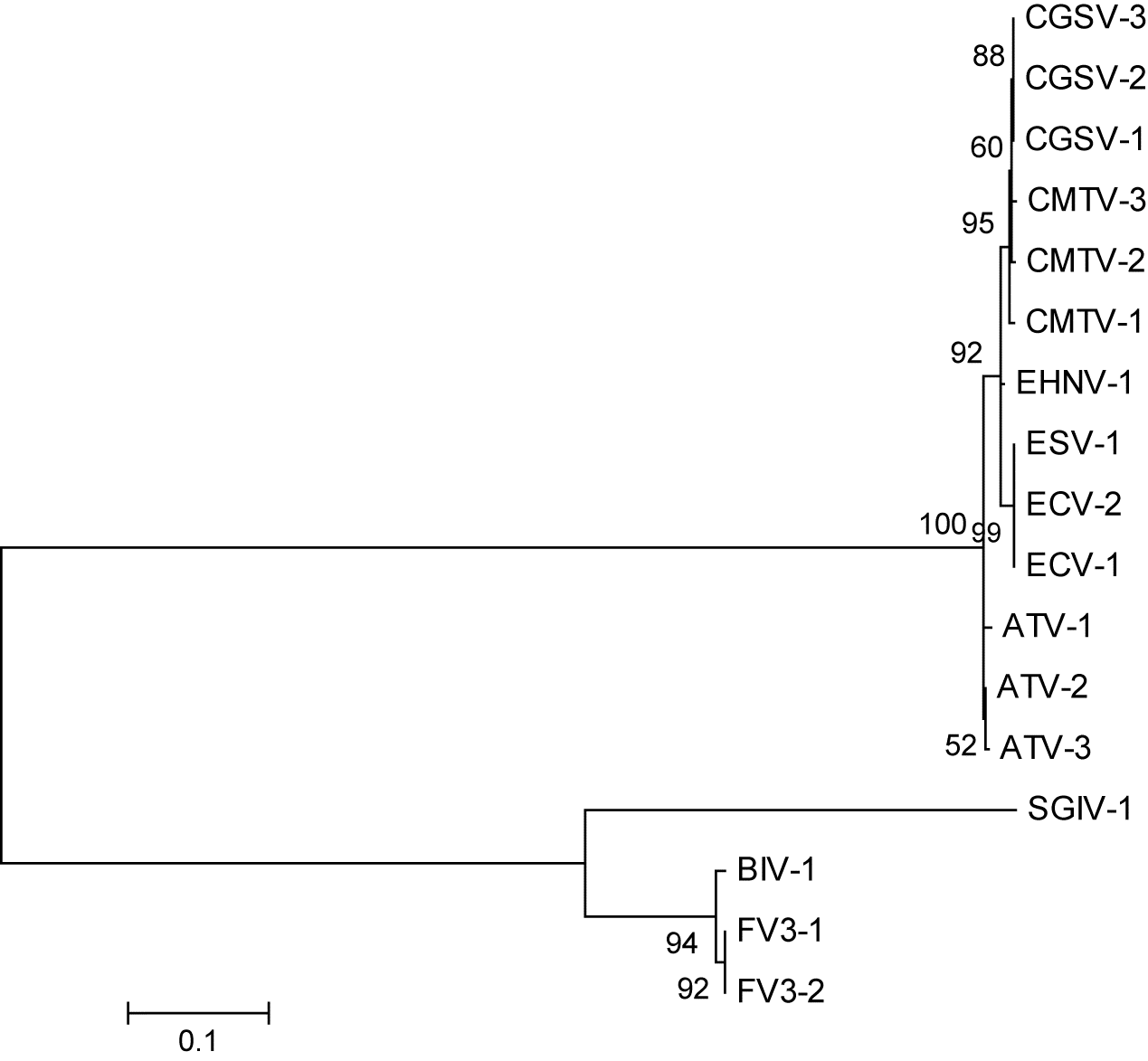
Fig. 3.
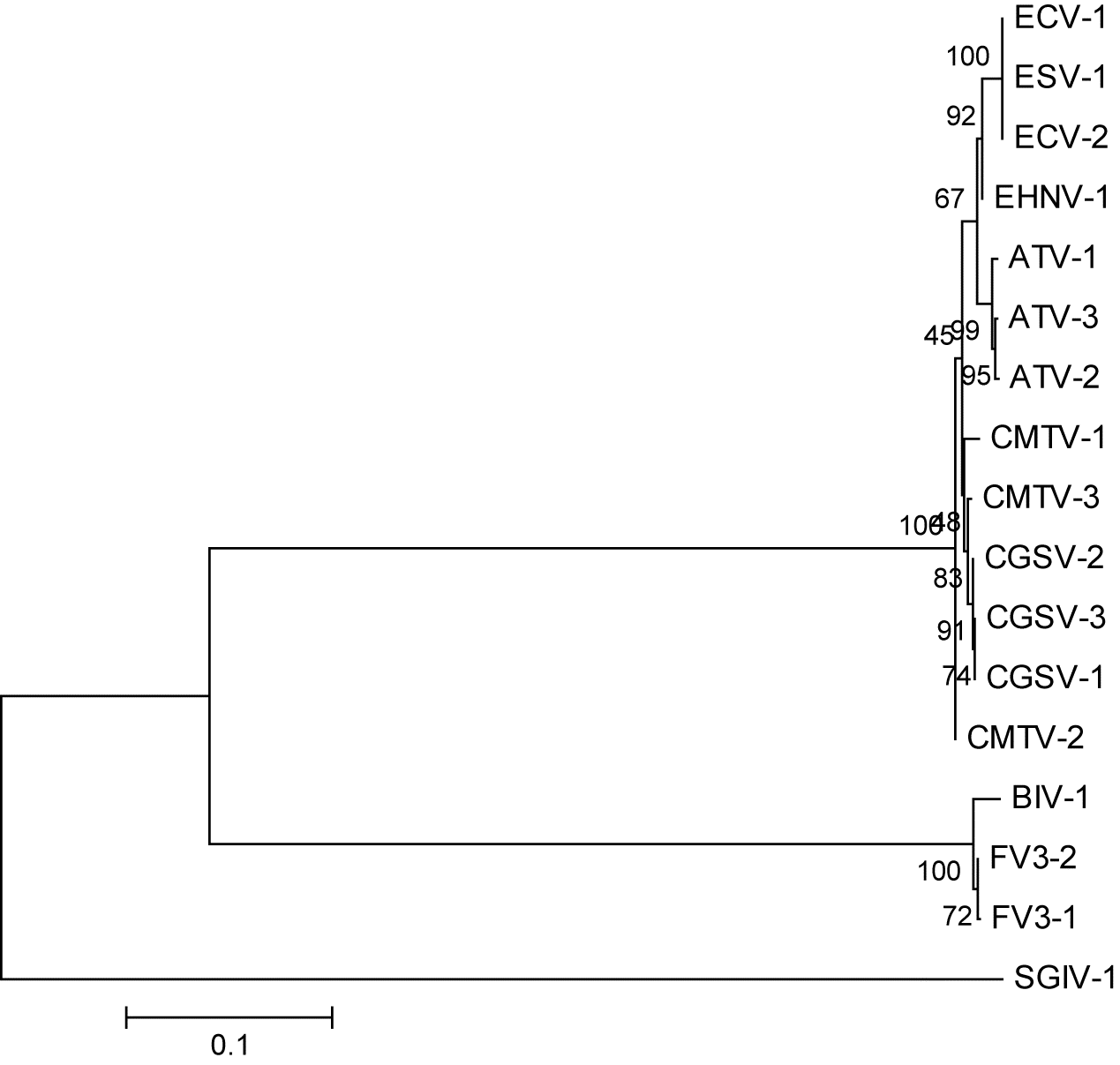
Fig. 4.
The trees that had the shortest symmetrical distance (i.e., 10 or below) to the MCP were made from ORFs 12L, 19R, 60R, 62L, 67L, 80L, 81R, 88R, 91R, and 94L. However, trees that had the shortest symmetrical distance to the full genomes were ORF 91R, ORF 94L, and ORF 95R (Table 2).
The best trees over all were made from ORF 19R, ORF 88R, ORF 91R (Fig. S19), and ORF 94L (Fig. S20) when all three types of analyses were considered together.
Dot-plot analysis of all 17 isolates of ranavirus used in the current study showed collinearity at the species level (e.g., Common midwife toad virus and Chinese Giant Salamander Virus, FV3, and Bohle Iridovirus) and there tends to only be smaller genomic rearrangements between most of the different species of Ranavirus. However, SGIV is not collinear with any other ranaviruses analyzed in this study (see Fig. 5).
Fig. 5.

Discussion
While most of the 24 core iridovirus genes used in this study will sort out the different ranavirus isolates to the species level, their utility is quite limited for finer-scale differentiation. There are only two genes that we feel make good approximations to phylogenetic trees constructed with the whole genome or the full sequence of the major capsid protein gene (ORF 90R in FV3), these are ORFs 19R and 88R when visually compared. ORF 19R is hypothesized to be a serine–threonine protein kinase (Eaton et al. 2007). This group of enzymes typically catalyze the transfer of phosphate groups from adenosine triphosphate (ATP) to proteins (Jacob et al. 2011). Our results here are surprising because viral serine–threonine protein kinases are usually highly conserved (Jacob et al. 2011). However, iridoviruses are hypothesized to have two serine–threonine protein kinases (ORF57R is a second FV3 serine–threonine kinase) in their core gene set (Eaton et al. 2007). It is common for genes that are duplicated in the genome to have one that is more variable than the other because of differences in selective pressures and mutation (see Shackelton and Holmes (2004) for a review of large DNA virus evolution). This is likely what is occurring in the case of ORFs 19R and 57R.
Open reading frame 88R is hypothesized to be an Erv I/Alr family protein (Eaton et al. 2007). These are sulfhydryl oxidase and are they are known to be far more divergent in viruses than in other organisms (Fass 2008). This sequence flexibility is likely what permits for a finer-scale differentiation between ranavirus species than other genes that are highly conserved based on functional needs. There is only a single gene that codes for Erv I/Alr family proteins in the core genes (Eaton et al. 2007), leaving out the potential for a gene duplication event and subsequent divergent evolution of the two genes within the iridovirus core genome.
Open reading frame 91R is hypothesized to be an immediate early protein infected-cell protein —46 homologue (Eaton et al. 2007). The function of ORF 91R has been partially characterized by Penny and Brunetti (2019). During infection, its protein product can be found in the nucleus and it is hypothesized that it is involved with viral DNA transcription as well as involved in one or more early viral replication processes (Penny and Brunetti 2019). Open reading frame 94L is hypothesized to be a homologue of a protein found in Clostridium tetani (Eaton et al. 2007). Its protein product localizes in the endoplasmic reticulum and are hypothesized to have a role in the viral modulation of cellular secretory pathways based on its structure (Penny and Brunetti 2019).
Our dot-plot analysis shows similar genomic rearrangements within ranaviral species. However, markedly different genomic arrangements can be seen between different species of ranavirus. Interestingly, SGIV shows minimal collinearity with the other ranavirus species examined (Fig. 5). The genome is very different in its arrangement, despite having the same core, and perhaps should be considered a different iridoviral genera instead of a species within the genus Ranavirus. In our phylogenetic analyses, SGIV tends also to be divergent, which supports this claim and previous reports of SGIV being the lowest percent similarity of all ranaviruses (see Stöhr et al. 2015).
Most of the 24 core iridovirus genes are able to more or less sort different ranavirus strains to the species level; however, it is highly likely that they are not able to show fine-scale differentiation (e.g., geographical or local adaptations), because they are not sufficiently polymorphic between species. Only two genes, ORFs 19R and 88R in FV3, make comparatively supported trees to the major capsid protein and full genomes visually. However, when the symmetrical distances are analyzed ORF 91R and 94L may be added to potentially informative sequences for phylogenetic analysis. We do recommend that single gene sequences alone not be used as there is evidence that multiple targeted concatenated genes are more useful in reconstructing the true phylogenies because of nucleotide shifts at the third position of the codon (e.g., yeasts, Collins et al. 2005).
Our dot-plot analysis suggests that each species of ranavirus has undergone unique genome rearrangements that are consistent at the species level. However, SGIV is highly divergent and may need to be considered a separate genera within the Iridoviridae instead of a species of ranavirus. Future directions should include determining the phylogenetic signal for each of the core genes and examining the utility of the more divergent core genes in sorting out local adaptations.
Acknowledgements
This work has been performed over several years and has been supported by various departments, including the Department of Biology (now the Department of Natural Sciences) and the Department of Mathematics and Computer Sciences. Special thanks to John C. George for supervising DRB’s research course and proofreading various versions of this manuscript. We would also like to thank the reviewers and editors for their comments on an earlier version of this manuscript as they have increased the quality of the document considerably.
References
Chinchar VG, Waltzek TB, and Subramaniam K. 2017. Ranaviruses and other members of the family Iridoviridae: their place in the virosphere. Virology, 511: 259–271.
Collins TM, Fedrigo O, and Naylor GJP. 2005. Choosing the best genes for the job: the case for stationary genes in genome-scale phylogenetics. Systematic Biology, 54(3): 493–500.
Duffus ALJ, and Andrews AM. 2013. Phylogenetic analysis of a frog virus 3–like ranavirus found at a site with recurrent mortality and morbidity events in southeastern Ontario, Canada: partial major capsid protein sequence alone is not sufficient for fine-scale differentiation. Journal of Wildlife Diseases, 49(2): 464–467.
Duffus ALJ, Waltzek TB, Stöhr AC, Allender MC, Gotesman M, Whittington RJ, et al. 2015. Distribution and host range of ranaviruses. In Ranaviruses: lethal pathogens of ectothermic vertebrates. Edited by MJ Gray and VG Chinchar. Springer, Cham, Switzerland. pp. 9–57 [online]: Available from link.springer.com/book/10.1007/978-3-319-13755-1.
Eaton HE, Metcalf J, Penny E, Tcherepanov V, Upton C, and Brunetti CR. 2007. Comparative genomic analysis of the family Iridoviridae: re-annotating and defining the core set of iridovirus genes. Virology Journal, 4(1): 11.
Eaton HE, Ring BA, and Brunetti CR. 2010. The genomic diversity and phylogenetic relationship in the family Iridoviridae. Viruses, 2(7): 1458–1475.
Fass D. 2008. The Erv family of sulfhydryl oxidases. Biochimica et Biophysica Acta (BBA)—Molecular Cell Research, 1783: 557–566.
Felsenstein J. 2008. TreeDist [online]: Available from evolution.genetics.washington.edu/phylip/doc/treedist.htm.
Fiorini N, Lefor V, Chevenet F, Berry V, and Arigon Chifolleau A-M. 2014. CompPhy: a web-based collaborative platform for comparing phylogenies. BMC Evolutionary Biology, 14: 253.
Geng Y, Wang KY, Zhou ZY, Li CW, Wang J, He M, et al. 2011. First report of a ranavirus associated with morbidity and mortality in farmed Chinese giant salamanders (Andrias davidianus). Journal of Comparative Pathology, 145(1): 95–102.
Granoff A, Came PE, and Rafferty KA Jr. 1965. The isolation and properties of viruses from Rana pipiens: their possible relationship to the renal adenocarcinoma of the leopard frog. Annals of the New York Academy of Sciences, 126(1): 237–255.
Jacob T, Van den Broeke C, and Favoreel HW. 2011. Viral serine/threonine protein kinases. Journal of Virology, 85(3): 1158–1173.
Jancovich JK, Qin Q, Zhang QY, and Chinchar VG. 2015. Ranavirus replication: molecular, cellular, and immunological events. In Ranaviruses: lethal pathogens of ectothermic vertebrates. Edited by MJ Gray and VG Chinchar. Springer, Cham, Switzerland. pp. 105–139.
Katoh K, Rozewicki J, and Yamada KD. 2019. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Briefings in Bioinformatics, 20(4): 1160–1166.
Penny E, and Brunetti CR. 2019. Localization of frog virus 3 conserved viral proteins 88R, 91R, and 94L. Viruses, 11: 276.
Price SJ, Garner TW, Nichols RA, Balloux F, Ayres C, de Alba AM, et al. 2014. Collapse of amphibian communities due to an introduced Ranavirus. Current Biology, 24(21): 2586–2591.
Qiu L, Chen MM, Wang RY, Wan XY, Li C, Zhang QL, et al. 2018. Complete genome sequence of shrimp hemocyte iridescent virus (SHIV) isolated from white leg shrimp, Litopenaeus vannamei. Archives of Virology, 163(3): 781–785.
R Core Team. 2019. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [online]: Available from R-project.org/.
Ridenhour BJ, and Storfer AT. 2008. Geographically variable selection in Ambystoma tigrinum virus (Iridoviridae) throughout the western USA. Journal of Evolutionary Biology, 21(4): 1151–1159.
Shackelton LA, and Holmes EC. 2004. The evolution of large DNA viruses: combining genomic information of viruses and their hosts. Trends in Microbiology, 12: 458–465.
Stöhr AC, López-Bueno A, Blahak S, Caeiro MF, Rosa GM, de Matos AP, et al. 2015. Phylogeny and differentiation of reptilian and amphibian ranaviruses detected in Europe. PLoS ONE, 10(2): e0118633.
Tamura K, Stecher G, Peterson D, Filipski A, and Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30(12): 2725–2729.
Teacher AGF, Cunningham AA, and Garner TWJ. 2010. Assessing the long-term impact of Ranavirus infection in wild common frog populations. Animal Conservation, 13(5): 514–522.
Tidona CA, Schnitzler P, Kehm R, and Darai G. 1998. Is the major capsid protein of iridoviruses a suitable target for the study of viral evolution? Virus Genes, 16(1): 59–66.
Waltzek TB, Miller DL, Gray MJ, Drecktrah B, Briggler JT, MacConnell B, et al. 2014. New disease records for hatchery-reared sturgeon. I. Expansion of frog virus 3 host range into Scaphirhynchus albus. Diseases of Aquatic Organisms, 111: 219–227.
Wright ES. 2016. Using DECIPHER v2.0 to analyze big biological sequence data in R. The R Journal, 8(1): 352–359.
Supplementary material
Supplementary Material 1 (DOCX / 72 KB)
- Download
- 71.99 KB
Information & Authors
Information
Published In

FACETS
Volume 5 • Number 1 • January 2020
Pages: 523 - 533
Editor: David Lesbarrères
History
Received: 19 February 2020
Accepted: 19 May 2020
Version of record online: 6 July 2020
Notes
This paper is part of a Collection titled “Ranavirus research: 10 years of global collaboration”
Copyright
© 2020 Ballard et al. This work is licensed under a Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Data Availability Statement
All relevant data are within the paper and Supplementary Material.
Key Words
Sections
Subjects
Authors
Author Contributions
ALJD conceived and designed the study.
All performed the experiments/collected the data.
All analyzed and interpreted the data.
ALJD drafted or revised the manuscript.
Competing Interests
The authors have declared that no competing interests exist.
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
D.R. Ballard, A.J. Davis, R.B. Fuller, A.R. Garner, A.D. Mileham, J.D. Serna, D.E. Brue, C.M. Harding, C.D. Dodgen, W. Culpepper, B. Piatt, S.E. Rosario, and A.L.J. Duffus. 2020. An examination of the Iridovirus core genes for reconstructing Ranavirus phylogenies. FACETS.
5(1): 523-533. https://doi.org/10.1139/facets-2020-0009
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. Advances on genomes studies of large DNA viruses in aquaculture: A minireview